#10 Updates - Telomere length estimation on MVP samples
Updates on running telseq and developing analysis methods
Compare telseq results from GCP and SGC cluster
-
Telomere length estimation has been completed for the subset of 10 CRAMS. We compared the results with the standalone study perfomed by Kruthika as a validation step. The results were the same for all 10 CRAMS between both runs.
-
We also looked at the runtime taken to run telseq on GCP vs SGC cluster.
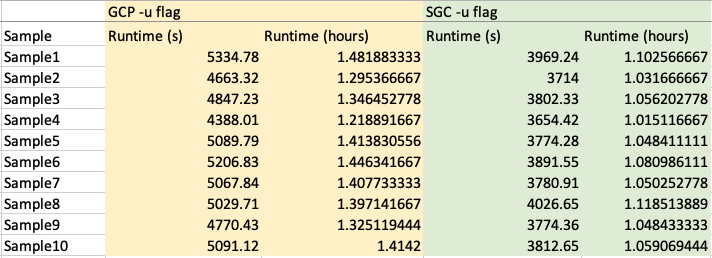
Running telseq with different parameters
After the previous discussion regarding the the read group, it was decided that telseq will be run using two different parameters. For both the runs the telseq.sh script was altered as required. All other parameters were kept the same when submitting the dsub job.
-
-u flag : This flag will tell telseq to treat all reads as if they were from the same bam file. The
-utag provided tosamtools viewis different from the-utag provided to telseq in this script. -
Remove
<RG>all together : Through this script, we remove the<RG>tag from the CRAM files because of which telseq is unable to identify read groups and hence all reads are treated as if they were from the same bam file.
In order to ensure that the outputs obtained from both the runs are the same, I performed a comparison as shown in the first part of this notebook). Since the outputs look the same, either flag can be used when scaling up the pipeline for 100 MVP samples.
Estimating cost and runtime for telseq runs
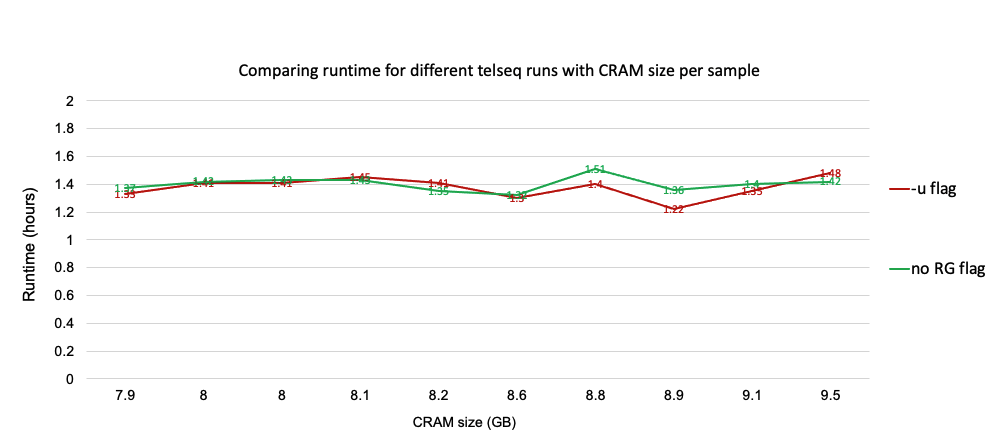
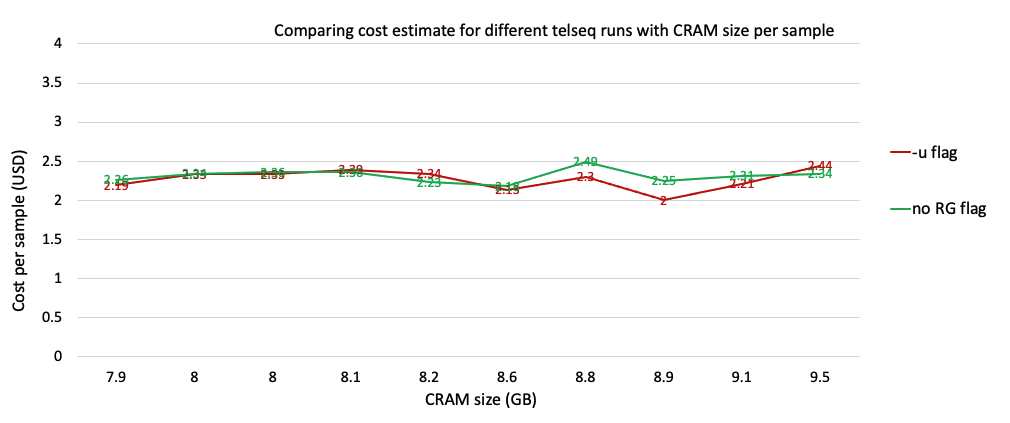
The runtime and cost are more or less the same for both telseq flags. For the next round, which is to scale up the pipeline for 100 MVP samples we will be using the -u flag.
Automatically estimating read length while running telseq
The read length is one of the parameters that is provided to telseq. After discussions, it was decided that it would be ideal to estimate this automaticatically instead of hardcoding the value. This has also been documented in this git issue. Although this is successful, for now we have decided to use the read length of 150 for scaling up the pipeline.
Running multiple telseq dsub jobs in parallel
While scaling up the pipeline, we need to submit multiple jobs in parallel which is also referred to as submitting a batch job. Instead of calling dsub repeatedly, we can create a tab-separated values (TSV) file containing the variables, inputs, and outputs for each task, and then call dsub once. The outcome is that we have a single job-id with multiple tasks. This way the tasks will be scheduled and run independently but the user can monitor and delete it as a group.
A snippet of the dsub script for telseq batch submission :
#!/bin/bash
dsub \
--provider google-v2 \
--project ${project_id} \
--regions us-west1 \
--machine-type n1-standard-8 \
--logging <PATH_TO_OUTPUT_LOGS> \
--name telseq_mvp \
--image jweinstk/telseq \
--task "<PATH_TO_TSV_FILE>" \
--script "<PATH_TO_INPUT_SCRIPT>"
A snippet of the TSV file containing variables, inputs, and outputs for each task:
--input CRAM --input REF --input REF_FAI --output TEXT
gs://PATH/TO/S1.cram gs://genomics-public-data/resources/broad/hg38/v0/Homo_sapiens_assembly38.fasta gs://genomics-public-data/resources/broad/hg38/v0/Homo_sapiens_assembly38.fasta.fai gs://PATH/TO/S1.txt
.
.
gs://PATH/TO/Sn.cram gs://genomics-public-data/resources/broad/hg38/v0/Homo_sapiens_assembly38.fasta gs://genomics-public-data/resources/broad/hg38/v0/Homo_sapiens_assembly38.fasta.fai gs://PATH/TO/Sn.txt
Analysing telseq output
Before scaling up the pipeline it would be ideal if we could understand and characterise the telseq output. In this notebook, I have visualized and represented a few values for the initial 10 samples that we have. I am still working on investigasting other methods of analysis/visualizations.
Currently all of the methods have been implemented on a set of 10 CRAMS. Once the whole pipeline is ready it will be scaled up to 100 CRAMS
Discussion
Discuss this update on our GitHub!