#43 From FastQ to gVCF: 5 Dollars and a Genome
Unraveling the mystery of variant calling in Trellis
Since beginning the process to make our system FedRAMP compliant, our team has been digging into the inner workings of Trellis. Trellis, developed by MVP alumnus Paul Billing-Ross is a database management system that is instrumental in processing our Whole Genome Sequencing (WGS) samples. Trellis not only tracks and makes data available through simple queries, but it automates the workflow that takes sequenced FastQ files to processed gVCF files.
In the last few months, it has become a priority to understand how this system and its architecture function. Daniel Cotter has been piecing together how the Trellis system is constructed and what changes need to be made to its architecture to make it both functional and compliant with FedRAMP regulations. I have been very interested in understanding how the variant calling pipeline works and what tools are needed to run it.
We have been left with some clues to help us piece these puzzles together. First, we have the Trellis paper that gives us some insights to the methodology involved in variant calling. Second, we have the source code implemented by Paul and Jina Song.
The publication mentions that we have incorporated the GTAK $5 genome analysis pipeline. It also gives a brief description of the variant calling process but omits many of the details involved in file processing. The most important information that can be extracted from this publication comes from the following:
Briefly, the human reference genome GRCh38 was used for mapping and variant calling. Alignment was performed by BWA-MEM (v0.7.15) with the command line: bwa mem -K 100,000,000 -p -v 3 -t 16 -Y $bash_ref_fasta. PCR duplicates were marked by Picard 2.15.0 and BQSR were performed in GATK 4.1.0.0, resulting in four-bin quality score compression. The alignment results in BAM were compressed to CRAM in a lossless manner. Variant calling was performed in GATK 4.1.0.0 using the haplotypeCaller function, resulting in gVCF files which contained reference call blocks, single nucleotide variants (SNVs), and short insertion and deletions (INDELs).
Additionally, the paper contains a relationship model (Fig. 1) that shows relationships formed in the variant calling process. While this model is helpful in understanding the flow of data through its relationships, many files are not represented. This most likely due to them not being stored to memory and never forming relationships in the database.
The information that we can derive from the paper gives us a good starting point. To obtain a full grasp of how variants are being called, however, the best plan of attack is to read through and document the source code. In the following sections, I will describe the script files stored on Git and attempt to describe the workflow in detail.
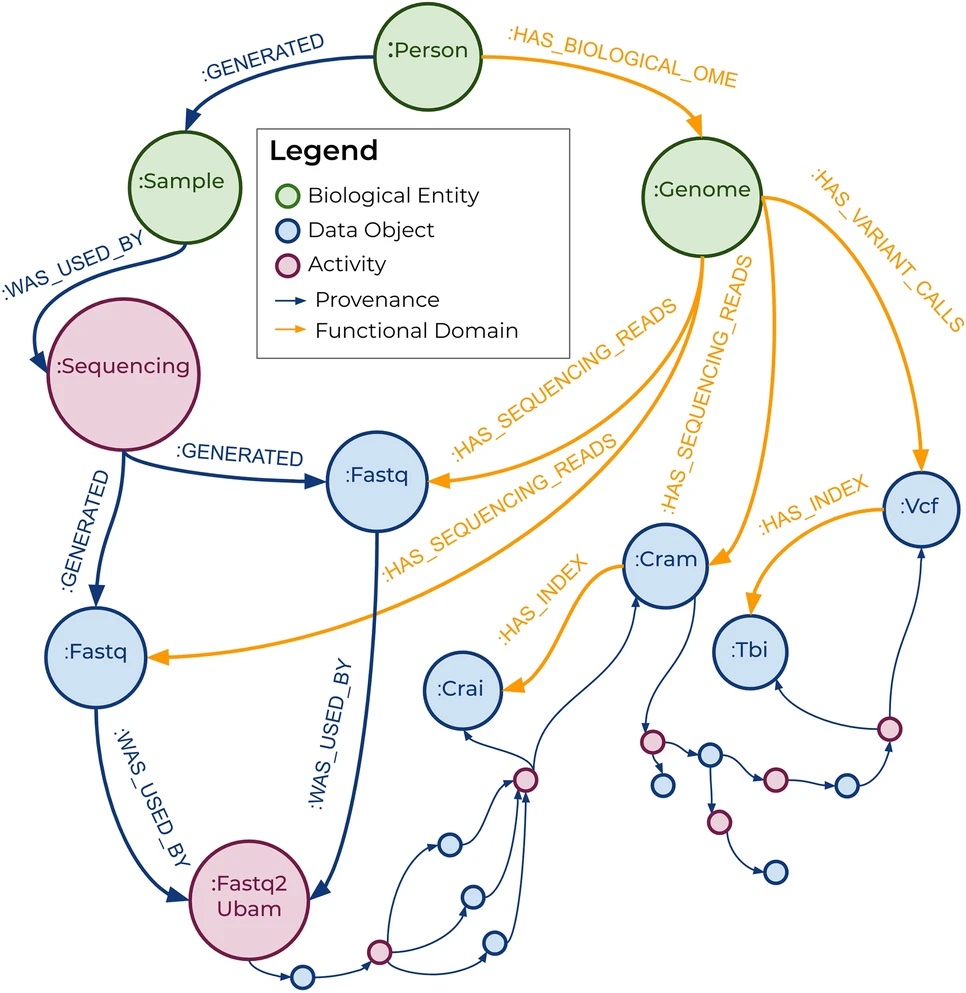 Fig 1. A relationship model showing the relationships formed in Trellis during the variant calling workflow.
Fig 1. A relationship model showing the relationships formed in Trellis during the variant calling workflow.
Files
Main files
The source code consists of scripts in several different file formats. The variant calling itself is carried out by tasks defined in WDL files that are imported and called in a WDL workflow. The workflow itself is submitted by a dsub script. The following describes these files and their functionality:
Sample level variable names are stored in a JSON file mvp.hg38.inputs.json.
The variant calling pipeline is initially submitted by the dsub command dsub_run_gatk
dsub_run_gatk calls a WDL file fc_germline_single_sample_workflow.wdl. This top level WDL file contains the defined workflow that calls tasks, which are defined in several WDL subfiles. The tasks defined in these WDL files are described below:
- GetBwaVersion: Gets current BWA version in use
- SamToFastqAndBwaMemAndMba: Convert uBAM to FastQ and map to the reference genome with BWA-MEM. The resulting BAM file is then remerged with the original uBAM using Picard’s MergeBamAlignment.
- SamSplitter: Splits a BAM file into smaller files. In this case a uBAM is split.
- SortSam: Sort and index mapped BAM files using SortSam.
- SortSamSpark: Spark implementation of SortSam that works on spark clusters using SortSamSpark. (Note: I do not believe that this has been implemented.)
- MarkDuplicates: Marks duplicates in mapped BAM files using MarkDuplicates.
- BaseRecalibrator: Recalibrates base quality scores of a mapped BAM file using BaseRecalibrator.
- ApplyBQSR: Apply recalibrated base scores from BaseRecalibrator to mapped BAMs using ApplyBQSR.
- GatherBqsrReports: Merge split BSQR reports into one file using GatherBqsrReports.
- GatherSortedBamFiles: Merge split, mapped, and sorted BAM files into one using GatherBamFiles.
- GatherUnsortedBamFiles: Merge unsorted BAM files into one using GatherBamFiles. (Note: used in split_large_readgroup)
- CheckContamination: Check contamination rate using VerifyBamID
germline_variant_discovery.wdl
- HaplotypeCaller_GATK35_GVCF: Call variants using the HaplotypeCaller walker in GATK version 3 (3.5?).
- HaplotypeCaller_GATK4_VCF: Call variants using the HaplotypeCaller walker in GATK version 4.
- MergeVCFs: Merge split gVCF files into one using MergeVcfs.
- HardFilterVcf: Filter variants with GATK’s VariantFilstration walker, using:
“QD < 2.0 || FS > 30.0 || SOR > 3.0 || MQ < 40.0 || MQRankSum < -3.0 || ReadPosRankSum < -3.0”. (Note: Only used when using GATK 4)
- CollectQualityYieldMetrics: Uses Picard’s CollectQualityYieldMetrics to collect metrics about reads that pass quality thresholds and Illumina-specific filters in uBAM files.
- CollectUnsortedReadgroupBamQualityMetrics: Uses Picard’s CollectMultipleMetrics on unsorted mapped BAMS to collect multiple classes of metrics running one or more of the metric collection modules.
- CollectReadgroupBamQualityMetrics: Collects multiple classes of metrics running one or more of the metrics collection modules, using Picard’s CollectMultipleMetrics, on sorted mapped BAMS.
- CollectAggregationMetrics: Collects multiple classes of metrics running one or more of the metrics collection modules, using Picard’s CollectMultipleMetrics, on aggregated mapped BAMS.
- CrossCheckFingerprints: Uses Picard’s CrossCheckFingerprints to check that all data in the set of input files, in this case BAMs, appear to come from the same individual. (only used in unmapped_bam_to_aligned.wdl)
- CheckFingerprint: Checks the sample identity of the sequence/genotype data in the provided BAM file against a set of known genotypes in a VCF file using Picard’s CheckFingerprint.(only used in unmapped_bam_to_aligned.wdl)
- CheckPreValidation: Checks whether the data has high duplication or chimerism rates.
- ValidateSamFile: Validates a BAM file using Picard’s ValidateSamFile.
- CollectWgsMetrics: Collect metrics about coverage and performance of a sorted aggregated BAM using Picard’s CollectWgsMetrics.
- CollectRawWgsMetrics: Collect whole genome sequencing-related metrics of a sorted aggregated BAM using Picard’s CollectRawWgsMetrics.
- CalculateReadGroupChecksum: Creates a hash code based on identifying information in the read groups (RG) of a BAM using Picard’s CalculateReadGroupChecksum. A checksum per RG is generated from the final BAM.
- ValidateGVCF: Validate a VCF file with a strict set of criteria described here. (I do not see this function used)
- CollectGvcfCallingMetrics: Collects per-sample and aggregate metrics from the provided VCF file using Picard’s CollectVariantCallingMetrics. (I do not see this function used.)
- This WDL file contains a workflow that is used to split uBAM files that are larger than a defined size cutoff. The workflow will split(scatter) the uBAM and proceed to convert and map the files using SamToFastqAndBwaMemAndMba.
- CreateSequenceGroupingTSV: Generates sets of intervals for scatter-gathering over chromosomes. Used to create list of sequences for scatter-gather parallelization in base recalibration.
- ScatterIntervalList: Calls picard’s IntervalListTools to scatter the input interval list into scatter_count sub interval lists. Variants are called on subinterval lists and merged.
- ConvertToCram: Converts a BAM file to CRAM using Samtools view.
- ConvertToBam: Converts a CRAM file to BAM using Samtools view.
- SumFloats: Calculates sum from a list of floats. This is used to estimate size of an aggregated BAM from split BAMs.
Additional files
The Git README notes that this pipeline requires samples to be un uBAM format before proceeding. Samples that are not already in uBAM file can have their FastQ files converted to uBAM using another dsub script: dsub_run_fastqtoubam. This script makes use of Picard’s FastqToSam.
There are two additional files that specify cloud resources.
- gatk-mvp-profile.sh: Grabs bucket and project information from Google Cloud.
- trellis-cloudbuild.yaml: Defines how files are copied to cloud storage.
The Pipeline
(Note: There are several splits and merges, before generating a final BAM, in the source code that will be described in this section. The source code mentions that these splits are carried out on sequenced reads across multiple flow cells for a single sample. The FastQ files that I have viewed on cloud storage are separated by lane on a single flow cell. However, this does not mean there are not some samples that span multiple flow cells. I do believe most of, if not all, the splits are done on lanes. But until I can affirm this, I will specify these splits as being done across flow cells, as specified in the source code.)
Typical variant calling pipelines that I have worked with in the past have been straightforward, in addition to following the Broad institute’s best practice standards (Fig. 2). An example of this includes the following steps.
- checking FastQ files for quality with FastQC
- trimming read adapters with a tool like Trimmomatic (or another adapter trimming tool) and re-evaluating their quality by running FastQC on the trimmed FastQ files
- aligning the read pairs to a reference genome with a tool like BWA-MEM
- marking duplicates and recalibrating base quality scores with Picard
- calling variants with Samtools or a GATK walker like HaplotypeCaller.
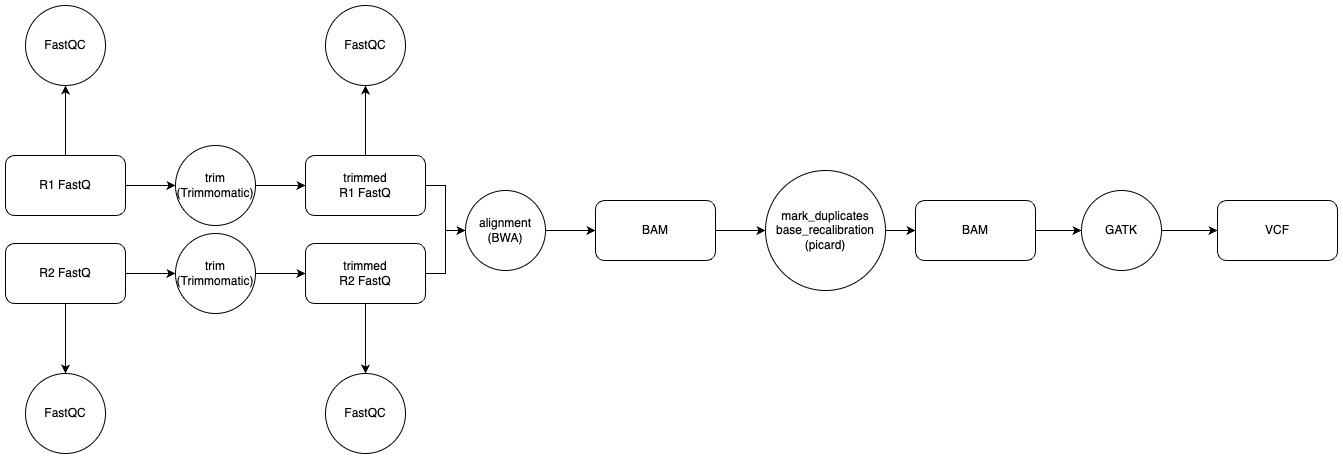 Fig 2. A typical pipeline incorporating GATK’s best practices represented in a workflow.
Fig 2. A typical pipeline incorporating GATK’s best practices represented in a workflow.
The pipeline implemented in Trellis is a bit more complicated (Fig. 3). The files delivered from Personalis are in FastQ format. The question of if we are receiving BCL files has been brought up in the past, and this does not appear to be the case. The FastQ files themselves are split by flow cell and converted into unmapped BAM files (uBAM). While not explicitly stated anywhere in the source code, this conversion is typically done to retain information that could possibly be lost when aligning directly from FastQ files. The uBAM files are scattered by flow cell prior to mapping and are processed in parallel. This is the first of four file scatters/splits. Two additional scatter steps are conducted during base recalibration and when applying recalibrated bases to the aligned BAM. Both of the latter scatter steps split per sequence groupings as defined by the CreateSequenceGroupingTSV task. The fourth scatter is conducted on intervals when calling variants with GATK, as defined by the ScatterIntervalList task. All four of these scatter steps were omitted from the workflow graph to reduce visual complexity. If a uBAM is larger than a predefined file size, it is scattered into smaller fragments to be processed in an additional scatter step. This is represented by the split_large_readgroup workflow step shown in the flow chart.
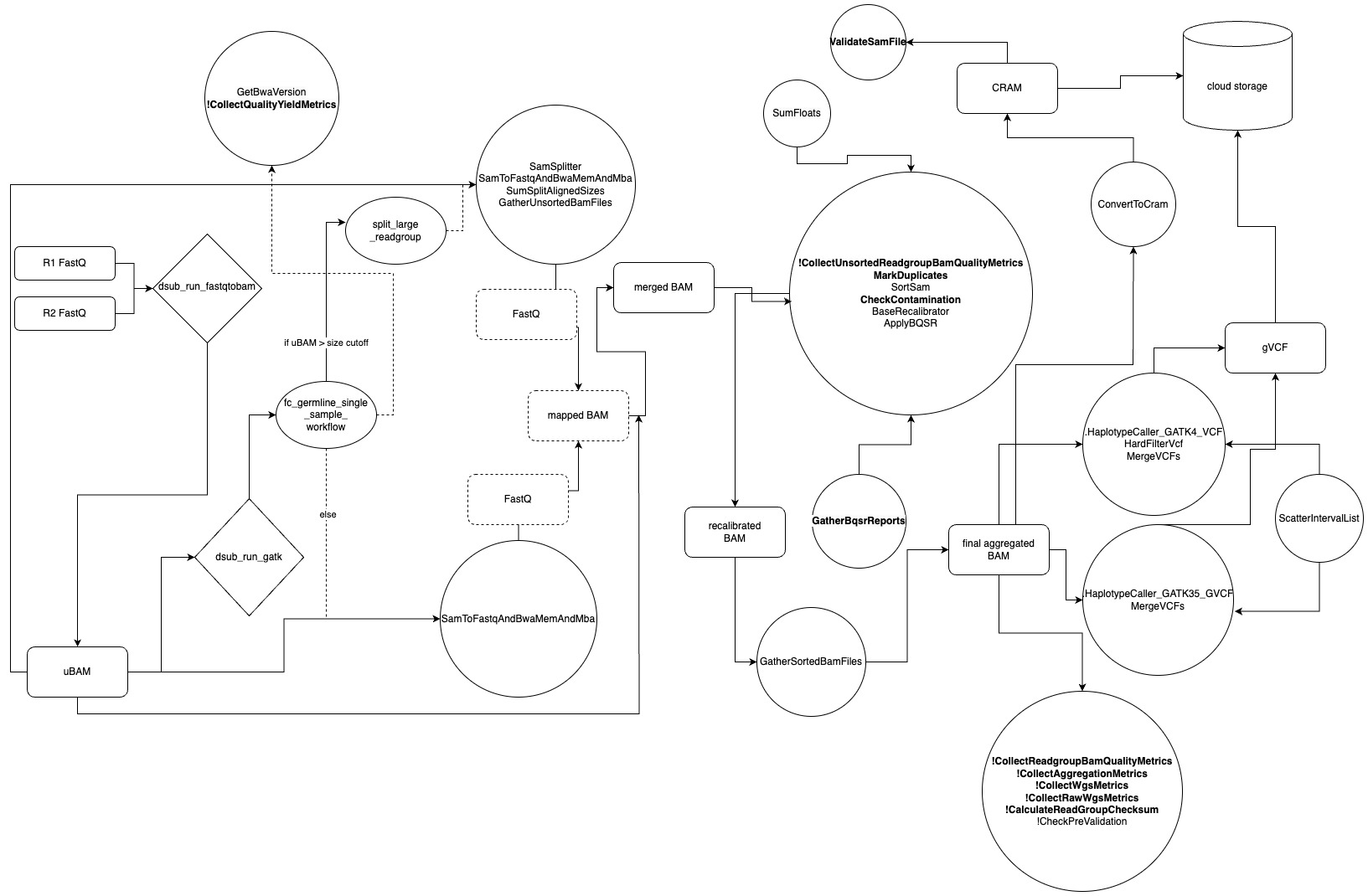 Fig 3. A workflow chart outlining the variant calling pipeline. Files, dusub commands, WDL workflows, WDL tasks, and cloud storage are represented by rectangles, diamonds, ovals, circles, and cylinders, respectively. Intermediate files not stored to memory are represented by perforated rectangles. Flow of data is represented by arrows, while WDL task calls are represented by perforated arrows. All WDL tasks, other than those called by the split_large_readgroup workflow, are called by the fc_germline_single_sample_workflow WDL workflow. Many of these connections have been omitted from the visualization to avoid a confusing cluster of arrows. Many WDL task need to be explicitly called by setting a setting a Boolean value to True. These are set to False by default and represented by an “!”. While all task logs and standard output are saved to cloud storage, some tasks generate additional files that are also transferred to cloud storage. These are shown in bold.
Fig 3. A workflow chart outlining the variant calling pipeline. Files, dusub commands, WDL workflows, WDL tasks, and cloud storage are represented by rectangles, diamonds, ovals, circles, and cylinders, respectively. Intermediate files not stored to memory are represented by perforated rectangles. Flow of data is represented by arrows, while WDL task calls are represented by perforated arrows. All WDL tasks, other than those called by the split_large_readgroup workflow, are called by the fc_germline_single_sample_workflow WDL workflow. Many of these connections have been omitted from the visualization to avoid a confusing cluster of arrows. Many WDL task need to be explicitly called by setting a setting a Boolean value to True. These are set to False by default and represented by an “!”. While all task logs and standard output are saved to cloud storage, some tasks generate additional files that are also transferred to cloud storage. These are shown in bold.
The workflow for variant calling is processed in the following steps:
- A mapped BAM file is generated within a single task. This task generates several files. It should be noted that none of these files are saved to cloud storage.
- First the uBAM is converted back into FastQ format using Picard’s SamtoFastq.
- The resulting FastQ is aligned with BWA-MEM with the following parameters:
bwa mem -K 100,000,000 -p -v 3 -t 16 -Y $bash_ref_fasta - The resulting BAM is merged with the original uBAM file using MergeBamAlignment.
- The mapped BAM files undergo duplicate marking and base recalibration.
- The recalibrated BAM is sorted and aggregated with other members of its RG. In this case the RG members should consist of sequences from the same sample across different flow cells.
- The final BAM is converted to a CRAM and copied to cloud storage.
- The final BAM is also used as the input to the HaplotypeCaller walker of GATK, and the resulting files are merged into a final gVCF that is saved to cloud storage.
Observations
While I was able to gain a lot of insight to the FastQ->gVCF process in Trellis by reviewing the scripts contained in the Git Repo, I was unable to locate some information. Two QC parameters that we use in sample filtering are FastQ quality results from fastQC and BAM alignment metrics from Samtool’s flagstat tool. Finding the source for running these tools is crucial, not just for processing new WGS data, but also for processing current WGS data in which this information is lacking.
As mentioned in the previous section, typical WGS variant calling workflows implement an adapter trimming step. A tool like Trimmomatic will trim sequence adapters, in addition to low quality bases. While it is not mandatory to complete this step, it is highly recommended. Current mapping tools do provide a level of trimming on the fly during the alignment step. For example, the current pipeline we are using makes use of the soft clipping feature of BWA-MEM. The results from our current data release shows that this may be sufficient for our data, but it is something to think about for the future. Especially at this time while we are overhauling Trellis for FedRAMP, and it has been recently noted that this pipeline has been deprecated by the Broad Institute.
Plans for the future
In looking deeper into our pipeline’s depreciation, I have found some interesting things. While I originally thought many of the scripts described in this blog were written in house, following the Broad’s best practices guidelines, it appears the WDL files are imported directly from the Broad’s $5 genome pipe Git repo. This would make our entire workflow deprecated and therefore no longer supported by the Broad Institute. This current pipeline was replaced in January of 2020 by a new pipeline. This new pipeline uses similar methodology upon first glance. However, this pipeline was also replaced in November of 2020. This would indicate that we are several versions behind.
The newest pipeline, warp, updates the older methodology but allows the option of using Dragen as an alternative pipeline. Moving to Dragen has recently become a discussion topic among our group. I have used Dragen tools in the past, not for WGS analysis but for viral variant calling and lineage analysis. For this project we used the Illumina BaseSpace cloud for data storage and processing. Other methods of running Dragen are available. Preliminary discussions have brought up the idea of using an Illumina server for our purposes. Strategies for incorporating this hardware into our FedRAMP approved architecture will need to be planned in this scenario. Running Dragen directly on our current cloud is another intriguing possibility. It seems that Dragen can be run on AWS and Azure, but I have not determined if it is compatible with Google Cloud.
Some questions that we will need to consider moving forward are:
- Whether we want to/when we want to update our pipeline?
- Will we re-run all of our already processed samples?
- How will we test/benchmark any new methodology before putting it into production?
- If we move to Dragen:
- will we need obtain a license from Illumina?
- will we need to update methodology for other projects? For example, The Dragen workflow indicates it outputs BAM/CRAM files. Would our telomere project need to also be updated with new CRAMs?
These discussions will need to continue as we decide the best strategy for moving forward with our future data releases.
Join the discussion here!