#50 Ambiguity in Gnomad BigQuery data
Background
In a previous study, our group compared the variants called in our latest data release with those from Gnomad’s version 3.0 release. During this analysis, we showed that there was a high number of variants that are uncommon between both studies. It was also shown that, when removing singletons, our data contained a higher number of uncommon variants per chromosome, signifying an increased probability of novel variant discovery among MVP researchers. While this data is very promising, Gnomad has since made several releases with new data and likely an increased number of variants. For our current publication being prepared, we will need to include the most up-to-date Gnomad data in this analysis.
Issues
The methodology involved in conducting these analyses incorporated BigQuery to manage data, taking advantage of both private MVP and public Gnomad BigQuery data. Gnomad has not uploaded any genomic release to BigQuery since version 3.0, which adds difficulty to our updates. While not on BigQuery, Gnomad’s release data is publicly available on Google Cloud in multiple formats. A Hail table of genomes is included among these formats. This is beneficial to us, as our data is stored as a Hail table, and we currently have methodologies in place to succeed in Hail.
Findings
I began by testing my methods with Gnomad’s version 3.0 Hail table to ensure there were no discrepancies with our BigQuery findings. While total variant counts per chromosome remained consistent, common variants between both data sets differed. While discouraging, I probed deeper to uncover why these discrepancies arose.
While our data contains a locus position specifying the site of the variant, I found that the Gnomad data in BigQuery contains both a start position and an end position, which presents some ambiguity. Each site’s start and end position differs, even among SNPs. I looked at the public Hail table and found it contained a similar single locus to our data. I also found that most sites matched the BigQuery end position.
In reviewing the source code for this analysis, I found that the end position is what was chosen as the position to match. To test my Hail methods, I created a new Hail table from the Gnomad BigQuery data, using the specified end positions as the locus. This test produced identical results to those of our original study.
Upon looking a bit deeper at the data, I found that the end position is not valid in insertions. The cause of this is that the end position accurately reflects the position of the last base in the variant. However, Hail uses the position of the first base as the locus address. It also became obvious that the start positions in Gnomad are not correct and should instead be the start position + 1. This would generate a start position equal to that of the end position in SNPs, reflecting the true start position for each variant.
I did not observe any deletions. I did, however, conclude that it is best to use the information in the Hail table as an accurate locus position to avoid any other abnormalities. This means that there are potentially fewer uncommon variants than we originally thought. After testing my Hail methods with end position data, I began to reanalyze the data to see how drastic the change is.
New results
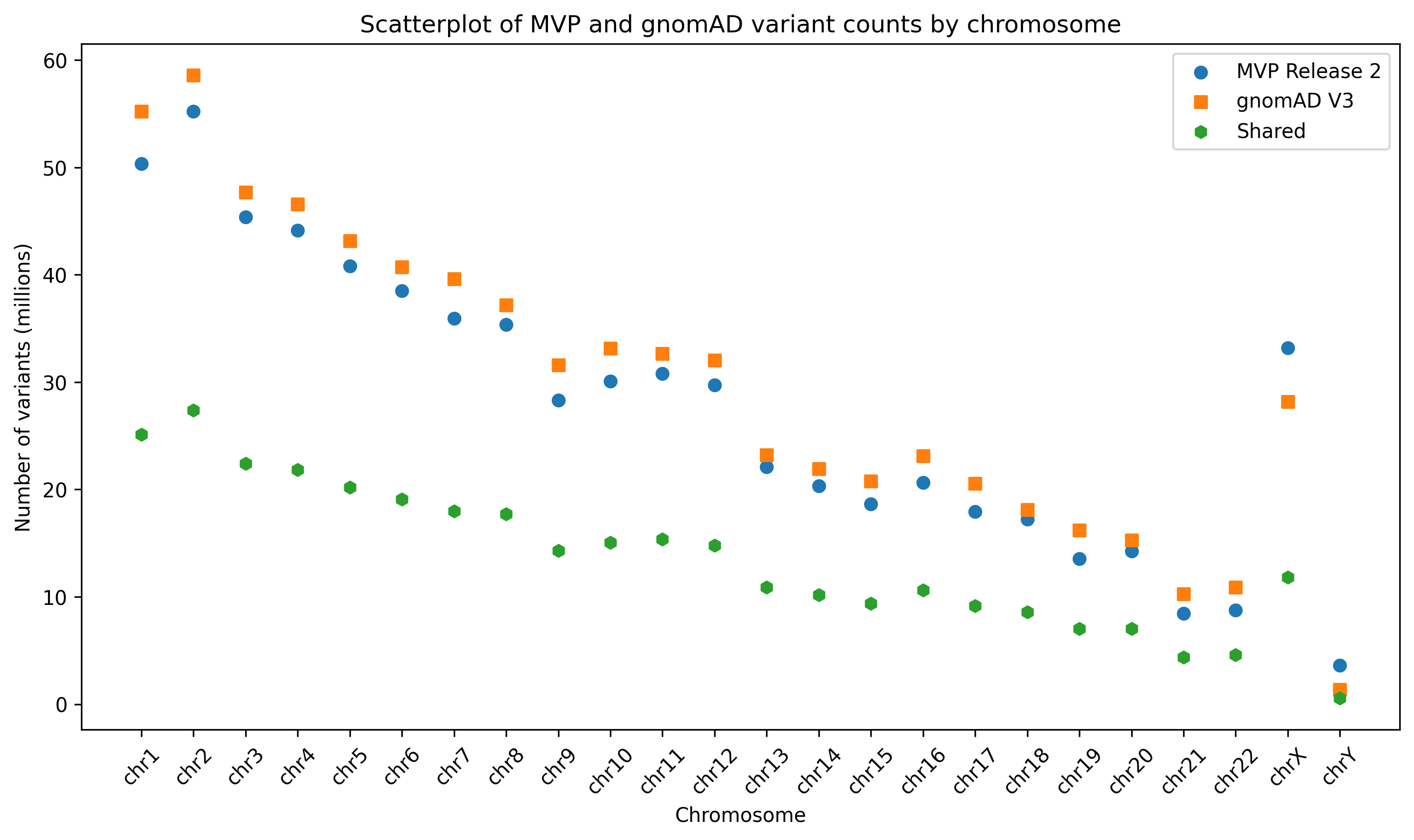
Reanalyzing the MVP to Gnomad v3.0 comparison using Hail showed a slight increase in shared variants per chromosome. These totaled approximately 18,000,000 new shared variants that we missed in the preliminary analysis (Table 1). However, this increase does not appear to present any significant changes from our original findings (Figure 1).
{kind=link}
| Chromosome | MVP variants | gnomAD variants | Previous Shared variants | New Shared Variants |
|---|---|---|---|---|
| All | 663,351,127 | 707,950,943 | 325,409,090 | 343,346,670 |
Table 1. Comparison of variant counts between MVP Release 2 and gnomAD v3, including updated results.
The number of shared variants will undoubtedly decrease when factoring out singletons, resulting in even less of a change. Tests using allele counts are currently being conducted, and I will update this post when these tests have been completed.
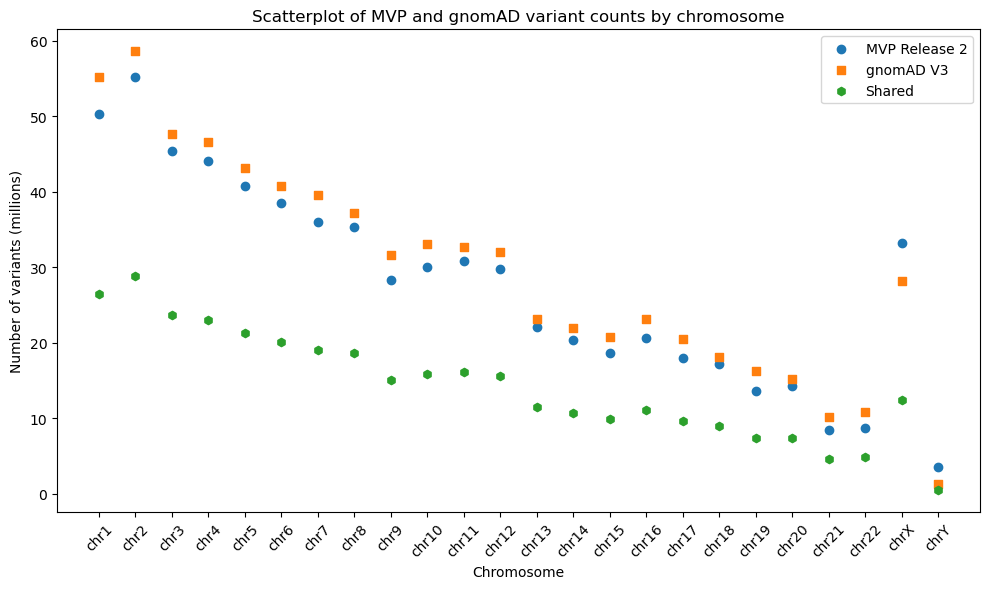 Figure 1. Reprocessed scatterplot of shared variant counts between MVP Release 2 and gnomAD v3, generated using Hail and stratified by chromosome.
Figure 1. Reprocessed scatterplot of shared variant counts between MVP Release 2 and gnomAD v3, generated using Hail and stratified by chromosome.
Future work
Now that I have established a workflow to analyze data in Hail format, I will need to repeat these analyses with the current Gnomad release data. One issue I have noticed is that Gnomad V4.1 was generated using a newer version of Hail. Because Hail does not allow for forward compatibility, we will need to upgrade our software to utilize this data. This is a good time to do this, as we revamp our entire structure for FedRAMP. However, I am hesitant to do this before publication. The reason for this is that our current workflow may also need updating to work with the new software version. This could delay any analysis we need done for revisions.
One workaround to this would be to use VCF files provided by Gnomad and convert them into a Hail table that is usable. Another solution is to upgrade our system and then downgrade if we need to quickly revise anything.
Join the discussion here!